The exact meaning of the term “retinitis” is inflammation of the retina. However, this collective term is not entirely correct today; many experts consider it outdated, since traditionally it includes a number of diseases that are not inherently infectious-inflammatory (for example, retinitis pigmentosa) and, strictly speaking, belong to the group of retinopathy, i.e. non-inflammatory pathologies. To avoid confusion, various classifications of retinitis are proposed, based, in particular, on the etiological, cause-and-effect principle.
General information about the disease
Retinal pigmentary degeneration has a low prevalence, on average affecting 1 in 5 thousand people. According to statistics, the disease more often affects men, although it is not gender specific.
Pathology is detected at the age of 20-30 years. Visual acuity decreases by 45-50 years and reaches 0.1.
Modern medicine is not able to completely overcome the disease and treatment comes down to the struggle to maintain visual acuity at an advanced stage.
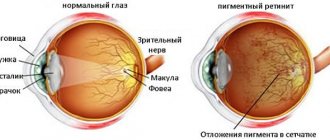
Retinitis pigmentosa is a genetic heterogeneous disease that appears as a result of disruption of the functioning of the retinal pigment epithelium with the development of other disorders.
The manifestation and severity of symptoms depend on the form of the disease. Pathology can manifest itself both at an early age and in adulthood and is entirely dependent on gene mutations.
Reference! Retinitis pigmentosa is inherited along any parental line. Children born in consanguineous marriages are at risk.
ICD-10 code
H35.5 Hereditary retinal dystrophies.
Forecast
It is impossible to make a long-term prognosis, since the degree of progression of the disease may vary. The foveal zone changes, causing a person's central vision to gradually decrease.
If the patient eats vitamins A and E every day, it is possible to stop the progression of rhinitis pigmentosa.
General forecast results:
- patients can easily read and write throughout their lives without glasses or surgery;
- 1/10 patients lose visual acuity by age 25;
- 1/10 – people become completely blind after 50.
Patients suffering from this disease should undergo regular examinations, because untimely treatment leads to the development of new diseases:
- the vitreous body changes;
- the optic nerve has drusen;
- development of myopia, glaucoma and keratoconus;
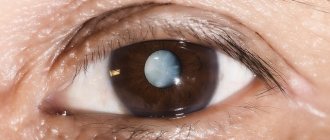
- the appearance of cataracts.
Causes
Any form of retinitis pigmentosa can be caused by one or more genes. Many mutations in the rhodopsin and peripherin genes cause a range of phenotypic manifestations of the disease.
The origin of retinitis pigmentosa is associated with metabolic disorders in photoreceptors and pigment epithelium. The brain stops receiving the necessary signals and this leads to the accumulation of toxic substances in the retina.
Molecular mutations in genes disrupt the production of rhodopsin (a precursor to retinol), which is vital for normal eye function.
Development ways:
- Gender dependence - transmitted from mother to son along with the X chromosome.
- Autosomal recessive - transmitted from two parents at once.
- Autosomal dominant - passed from one parent.
- Not hereditary - very rare. Usually the disease is passed on from generation to generation.
Types of retinitis pigmentosa
Based on the fact that retinitis pigmentosa develops at the genetic level, scientists identify the following types of pathology:
- Autosomal dominant. Recorded as the most common type of deviation. His treatment has a positive effect.
- Autosomal recessive. May occur with complications. According to statistics, it is in second place in terms of detection frequency.
- Linked to the X chromosome. It is detected in rare cases. Very difficult to treat.
Retinitis pigmentosa is also divided into typical and atypical disease. The first one hits the stick system. The second is divided into the following forms of the disease:
- Pigmentless. It occurs without accumulation of pigment substances in the fundus.
- Sectoral horseshoe. The pigment accumulated in the fundus is distributed in the shape of a horseshoe in the inferotemporal region. Characterized by long-term symptoms.
- Inverted or central. The pigment accumulates in the cone region and in the central zone of the retina. At the same time, visual acuity drops sharply.
- White spot. A large number of white pigment spots form in the fundus.
- External hemorrhagic or Coats' retinitis. The disease occurs with changes in the choroid of the retina.
Retinitis pigmentosa is a disorder inherited from either parent. The risk of its formation increases if relatives enter into marriage. What is retinitis pigmentosa and its main characteristics are described here:
Classification
The modern classification of the disease depends on the mechanism of hereditary transmission of the genetic disorder:
- autosomal dominant inheritance
- autosomal recessive inheritance
- X-linked inheritance mechanism
- caused by mitochondrial DNA mutations
Based on the nature of the disease and the affected areas, typical and atypical forms are distinguished.
There are also classifications according to the presence or absence of concomitant abnormalities and according to the age of manifestation of the pathology (congenital, juvenile).
Koat's retinitis is a pathological development of the blood vessels of the eye. The disease is accompanied by bleeding from the affected vessels in the posterior wall of the retina, and against the background of increased cholesterol levels, it eventually causes retinal detachment.
Rheumatic retinovasculitis - appears as a white formation located at the location of the vessels. With late diagnosis, it results in a stable loss of visual acuity and the development of secondary perivascular fibrosis.
Toxoplasmic retinitis is manifested by the formation of scarring and atrophic changes without an inflammatory process. Accompanied by clouding of the vitreous body.
Syphilitic choriopetinitis - causes swelling of the retina and nerve, hemorrhage and opacification of the vitreous body.
Cytomegalovirus retinitis - affects HIV-infected people, is infectious in nature, and is complicated by retinal detachment.
Viral retinitis - caused by influenza, adenovirus and herpes viruses, manifests itself in clouding of the retina. The pathology is eliminated on its own, and by restoring the ability to clearly perceive objects, a relapse is possible.
Septic retinitis is accompanied by damage to the retinal vessels. Caused by pyogenic bacteria in the patient's body. The progression of the anomaly causes swelling of the eye nerve, vitreous opacification and panophthalmitis.
Typical
Visual acuity (central and peripheral) with this form remains normal for a long time. However, at this time the rod system is damaged and pockets of pigment accumulation appear.
Atypical
Atypical retinitis, depending on the affected areas of the retina and the nature of the progression of the pathology, is divided into:
- central
- non-pigmented
- white-spotted
- pseudopigmentous
- generalized hereditary retinal dystrophy associated with systemic diseases and metabolic disorders.
conclusions
- Retinitis pigmentosa is an inherited vision disorder. The reason for its appearance is the incorrect formation of the gene.
- There are 4 types of disease depending on the affected chromosome.
- In the early stages, signs of pathology are almost invisible, so the disease can manifest itself at any age.
- Retinitis pigmentosa cannot be completely eliminated. But it is quite possible to slow down the processes of vision loss. If you ignore the symptoms and do not contact an ophthalmologist, the patient risks completely losing the ability to see. Find out about female color blindness in this material.
Symptoms
Retinal pigmentary degeneration manifests itself in adolescence; the dominant type can manifest itself at a more mature age.
The first symptom is a long adaptation of vision to poor lighting.
Further development of the disease causes the following symptoms:
- Periodic decrease in visual acuity.
- Disorientation in space and darkness.
- Difficult adaptation when moving from light to dark and vice versa.
- Good vision in daylight and blindness in the dark.
- Narrowing of peripheral vision.
- Distortion of the outlines of objects.
- Formation of blind spots in the field of view.
- Tunnel vision (narrowing of the visual field).
- Immunity to shades of blue.
Attention! Gene mutations affect the condition of the blood vessels of the eyes, this causes the destruction of cones, clouding of the lens, thinning of the sclera, and retinal edema.
Clinical triad
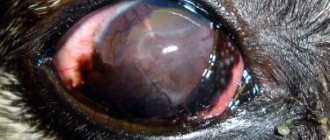
During ophthalmoscopy, a triad of symptoms is observed:
- bone bodies are typical foci of retinal hyperpigmentation;
- narrowing and atrophic changes in the fundus arterioles;
- pallor of the optic disc (waxy appearance)
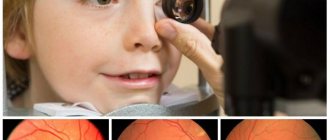
Diagnostics
To make a diagnosis, the doctor needs to know which gene led to the onset of the disease and the distribution of such mutation in the family tree.
The diagnosis of retinitis pigmentosa is established in the presence of specific defects in the vitreous body.
Important! Children can be diagnosed with this after 6 years of age, when the child’s visual organs are fully formed.
Pathology research methods:
- taking anamnesis of close relatives
- examination of the fundus with dilated pupils and multiple magnification
- color perception testing
- assessment of the condition of eye vessels using fluorescein angiography
- determining the degree of adaptation to light and darkness using electroretinography
- Electrooculography – determination of the amplitude of vision in both eyes
- CT scan
- molecular genetic tests
Treatment and prognosis
Retinitis pigmentosa is a little-studied disease that cannot be treated, but there is a chance of not aggravating the condition and preserving vision.
Lack of treatment can cause the development of concomitant eye diseases (cataracts, keratoconus, etc.).
Measures to maintain eye health:
- Preparations for maintaining vision and protecting the eyes - lutein, zeaxanthin, lycopene, blueberry extract.
- Strengthening the vascular wall (rutin).
- Low cholesterol diet.
- Diuretics for retinal edema.
- Vasodilators for better oxygen supply to the organs of vision.
Modern science is working to develop new treatment methods:
- Transplantation of cells responsible for pigmentation. Defective cells are replaced with healthy ones, as a result of which genes and chromosomes become in the correct sequence.
- Retinal replacement helps to significantly improve the condition, but does not completely cure the disease.
- Implantation – transplantation of donor stem cells and tissues (a method under active development).
In general, the prognosis is unfavorable; supportive treatment can delay the onset of blindness by 5-10 years.
Useful video
How does a person with retinitis pigmentosa see:
Notes
- ↑ 1 2
Genetic Reactivation of Cone Photoreceptors Restores Visual Responses in Retinitis pigmentosa
(undefined)
. - Koenekoop, R. K.;
Loyer, Magali; Hand, Collette K; Al Mahdi, Huda; Dembinska, Olga; Beneish, Raquel; Racine, Julie; Rouleau, Guy A. Novel RPGR mutations with distinct retinitis pigmentosa phenotypes in French-Canadian families // American Journal of Ophthalmology (English) Russian. : journal. - 2003. - Vol. 136, no. 4. - P. 678-668. - doi:10.1016/S0002-9394 (03)00331-3. - Farrar GJ, Kenna PF, Humphries P.
On the genetics of retinitis pigmentosa and on mutation-independent approaches to therapeutic intervention (English) // The EMBO Journal (English) Russian. : journal. - 2002. - March (vol. 21, no. 5). — P. 857—864. - doi:10.1093/emboj/21.5.857. - PMID 11867514. - Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet 2013:84:132–141.
- ↑ 1 2 Hartong DT, Berson EL, Dryja TP
Retinitis pigmentosa (English) // The Lancet. - Elsevier, 2006. - November (vol. 368, no. 9549). - P. 1795-1809. - doi:10.1016/S0140-6736 (06)69740-7. - PMID 17113430. - OMIM 268000
- ↑ 1 2 Berson EL, Rosner B., Sandberg MA, Dryja TP
Ocular findings in patients with autosomal dominant retinitis pigmentosa and a rhodopsin gene defect (Pro-23-His) // JAMA Ophthalmology (English) Russian. : journal. - 1991. - January (vol. 109, no. 1). - P. 92-101. - doi:10.1001/archopht.1991.01080010094039. - PMID 1987956. - Senin II, Bosch L., Ramon E., et al.
Ca2+/recoverin dependent regulation of phosphorylation of the rhodopsin mutant R135L associated with retinitis pigmentosa (English) // Biochemical and Biophysical Research Communications (English) Russian. : journal. - 2006. - October (vol. 349, no. 1). - P. 345-352. - doi:10.1016/j.bbrc.2006.08.048. - PMID 16934219. - Dryja TP, McGee TL, Reichel E, et al.
A point mutation of the rhodopsin gene in one form of retinitis pigmentosa (English) // Nature: journal. - 1990. - January (vol. 343, no. 6256). — P. 364—366. - doi:10.1038/343364a0. - PMID 2137202. - Dryja TP, McGee TL, Hahn LB, et al.
Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa (English) // The New England Journal of Medicine: journal. - 1990. - November (vol. 323, no. 19). — P. 1302—1307. - doi:10.1056/NEJM199011083231903. - PMID 2215617. - Berson EL, Rosner B., Sandberg MA, Weigel-DiFranco C., Dryja TP
Ocular findings in patients with autosomal dominant retinitis pigmentosa and rhodopsin, proline-347-leucine // American Journal of Ophthalmology (English)russian . : journal. - 1991. - May (vol. 111, no. 5). - P. 614-623. - PMID 2021172. - Inglehearn CF, Bashir R., Lester DH, Jay M., Bird AC, Bhattacharya SS
A 3-bp deletion in the rhodopsin gene in a family with autosomal dominant retinitis pigmentosa (English) // American Journal of Human Genetics (English). )Russian : journal. - 1991. - January (vol. 48, no. 1). - P. 26-30. - PMID 1985460. - Oh, Kean T.;
Weleber, R. G.; Lotery, A; Oh, DM; Billingslea, AM; Stone, EM Description of a New Mutation in Rhodopsin, Pro23Ala, and Comparison With Electroretinographic and Clinical Characteristics of the Pro23His Mutation // JAMA Ophthalmology (English) Russian. : journal. - 2000. - 1 September (vol. 118, no. 9). — P. 1269—1276. - doi:10.1001/archopht.118.9.1269. - PMID 10980774. - Berson, Eliot L.;
Rosner, B; Sandberg, M.A.; Hayes, K. C.; Nicholson, B. W.; Weigel-DiFranco, C; Willett, W. A Randomized Trial of Vitamin A and Vitamin E Supplementation for Retinitis Pigmentosa // JAMA Ophthalmology (English) Russian. : journal. - 1993. - June 1 (vol. 111, no. 6). - P. 761-772. - doi:10.1001/archopht.1993.01090060049022. - PMID 8512476. - Berson EL
Long-term visual prognoses in patients with retinitis pigmentosa: the Ludwig von Sallmann lecture (English) // Exp. Eye Res. : journal. - 2007. - Vol. 85, no. 1. - P. 7-14. - doi:10.1016/j.exer.2007.03.001. — PMID 17531222.This is not verified by many Doctors - Humayun, M.S.;
Dorn, J.D.; da Cruz, L; Dagnelie, G; Sahel, J.A.; Stanga, P.E.; Cideciyan, A. V.; Duncan, J.L.; Eliott, D; Filley, E; Ho, A.C.; Santos, A; Safran, AB; Arditi, A; Del Priore, L.V.; Greenberg, R. J.; Argus II Study, Group. Interim results from the international trial of Second Sight's visual prosthesis (English) // Ophthalmology : journal. — 2012. — April (vol. 119, no. 4). - P. 779-788. - doi:10.1016/j.ophtha.2011.09.028. - PMID 22244176. - FDA approves first retinal implant for rare eye disease (undefined)
. Reuters (February 14, 2013). Access date: February 14, 2013. - FDA approves first retinal implant for adults with rare genetic eye disease (unspecified)
(inaccessible link). Food and Drug Administration (February 14, 2013). Access date: January 5, 2015. Archived December 23, 2014. - The blind may soon see again as science prepares to market high-tech cyborg eye (undefined)
.
Daily Mail
(9 February 2013). Access date: February 12, 2013. - 'First Bionic Eye' Retinal Chip for Blind (June 29, 2013). Retrieved June 30, 2013.
- Rush University Medical Center (2005-01-31). Ophthalmologists Implant Five Patients with Artificial Silicon Retina Microchip To Treat Vision Loss from Retinitis Pigmentosa
. Press release. Archived from the original on February 8, 2005. Retrieved 2007-06-16. - MacLaren, R. E.;
R. A. Pearson; A MacNeil; RH Douglas; T. E. Salt; M Akimoto; A Swaroop; J. C. Sowden; RR Ali. Retinal repair by transplantation of photoreceptor precursors (English) // Nature (journal) : journal. - 2006. - November 9 (vol. 444, no. 7116). - P. 203-207. - doi:10.1038/nature05161. - PMID 17093405. - Sato S., Omori Y., Katoh K., et al.
Pikachurin, a dystroglycan ligand, is essential for photoreceptor ribbon synapse formation (English) // Nat. Neurosci. : journal. - 2008. - August (vol. 11, no. 8). — P. 923—931. - doi:10.1038/nn.2160. - PMID 18641643. - Lightning-Fast Vision Protein Named After Pikachu July 24, 2008
- Experiments show retinitis pigmentosa is treatable December 22, 2012
- OASIS
- THC May Slow Vision Loss From Retinitis Pigmentosa
- Restoring Visual Function to Blind Mice with a Photoswitch that Exploits Electrophysiological Remodeling of Retinal Ganglion Cells: Neuron
- Neil Fachie https://www.paralympics.org.uk/gb/athletes/neil-fachie Archived August 31, 2012 on the Wayback Machine
- McDonald, Margie
. Wheel turns a full circle as proud Lindy rides for two countries in Beijing, The Australian (May 31, 2008), p. 54. Accessed February 1, 2012. - CSI Cast: Jon Wellner (unspecified)
. CBS. Access date: October 5, 2010. - Paumgarten, Nick
Doh!
Dept: The $40-Million Elbow (unspecified)
. The New Yorker. Access date: August 13, 2012. - Interview with Metro Online (unspecified)
. Access date: September 4, 2006.